이 동영상을 재생하려면 YouTube의 쿠키 및 기타 외부 서비스를 허용해 주세요.
이 동영상을 재생하려면 YouTube의 쿠키 및 기타 외부 서비스를 허용해 주세요.
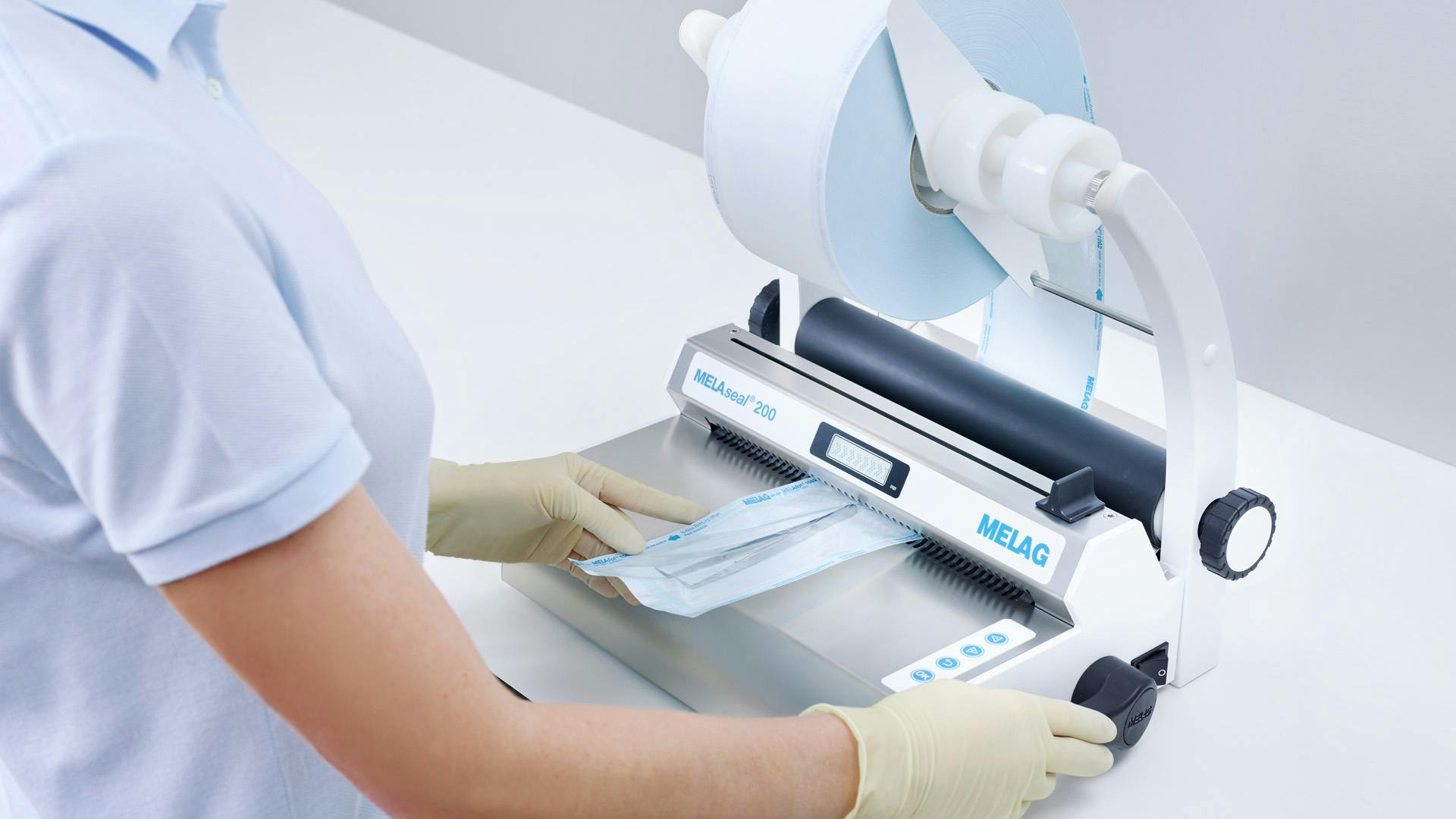
작업을 시작하기 전에 하루에 한 번씩 육안 및 기계 점검을 통해 테스트 밀봉 이음새를 수행 및 점검합니다. 박리 테스트를 수행하려면 밀봉된 필름 포장을 오토클레이브에서 멸균합니다. 증기 멸균기에서 꺼낸 후 밀봉 이음새를 박리 방향으로 천천히 떼어냅니다. 필름이 종이에서 완전히 벗겨졌는지 육안으로 확인합니다. 필름 포장을 개봉할 때 10mm 이상 닳아서는 안 됩니다.
박리 테스트 결과와 모든 추가 일상 점검을 검증 가능한 방식으로 문서화합니다. 이를 위해 멜라그 템플릿 체크리스트를 제공합니다.
매일 씰 이음새의 씰 점검은 MELAG 씰 점검으로 수행할 수 있습니다. 멜라그 씰 체크는 안전하고 효율적인 방법으로 씰 이음새를 확인할 수 있습니다. 인쇄 샘플의 대비를 통해 실무팀은 필름 층이 종이 면과 올바르게 융합되었는지 여부를 빠르고 쉽게 확인할 수 있습니다. 씰 체크는 멸균 파우치에 삽입되며, 전체 씰 이음새에 변색이 없고 윤곽이 선명하면 성공적인 것입니다. 씰링 장치의 일상적인 테스트에 대한 추적 가능한 문서화를 위해 MELAconnect 앱을 제공합니다. 이 앱을 사용하면 스마트폰이나 태블릿의 카메라를 통해 씰 검사를 쉽게 보관할 수 있습니다.
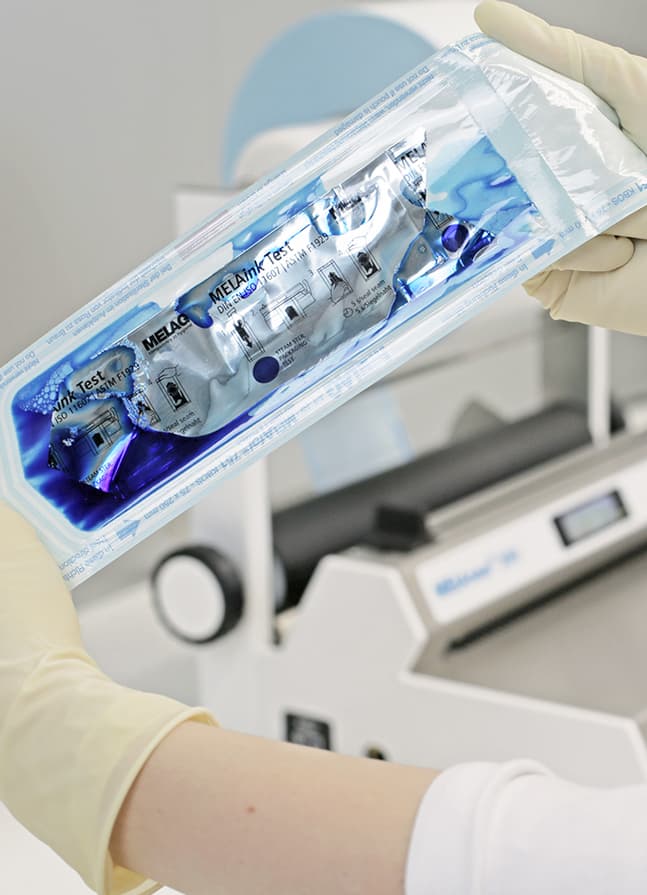
매주 실시하는 MELAink 테스트를 통해 씰 이음새의 불규칙성을 더욱 정확하게 파악할 수 있습니다. 잉크 테스트는 투명한 멸균 패키지에 넣고 밀봉합니다. 그런 다음 테스트 백에서 씰 이음새 방향으로 잉크를 밀어냅니다. 잉크가 퍼지면 포장의 네 면을 모두 테스트하여 밀봉 이음새의 불규칙성, 결함 또는 채널이 있는지 확인합니다.
씰링 심 안정성 테스트는 매년 씰링 심의 인장 강도(인열 저항)를 점검하고 MELAG 씰링 장치를 사용한 포장 공정의 유효성을 검증하는 데 사용됩니다. 씰 심 안정성 테스트는 국제 표준에 따라 씰링 장치의 성능 검증을 위한 표준화된 테스트 절차입니다. 테스트는 유효한 결과를 보장하기 위해 첨단 전자 테스트 기계로 수행됩니다. 멜라그 씰링 심 안정성 테스트 신청서를 다운로드하세요. 이 양식에는 씰 심 안정성 테스트를 수행하는 데 필요한 모든 단계가 설명되어 있습니다.
씰링 장치는 멸균된 기구를 환자 치료에 필요할 때까지 오염 없이 보관할 수 있게 해줍니다. 포장 밀봉 심의 신뢰성과 안전성을 확인하기 위해 "밀봉 심 안정성 테스트" 또는 MELAink 테스트와 같은 일상적인 테스트를 제공합니다. 이러한 테스트는 지역 당국에서 정한 간격으로 수행해야 합니다.
병원 부문에서는 오랫동안 씰링 절차 중에 온도, 접촉 압력, 씰링 시간의 세 가지 프로세스 파라미터를 문서화하여 자동 점검을 수행하는 씰링 장치를 사용해 왔습니다. 현재 많은 국가에서 실무 기반 의료 분야에서 이러한 검증 가능한 밀봉 장치의 사용은 의무 사항은 아니지만 권장 사항인 경우가 많습니다.
그럼에도 불구하고 신규 구매를 고려하는 병원과 클리닉은 모든 공정 파라미터를 지속적으로 모니터링하고 문서화하여 재현 가능한 안전성을 보장하는 유효성 검사 가능한 밀봉 장치를 선택하는 것을 고려해야 합니다.
멜라그의 씰링 장치 MELAseal Pro와 MELAseal 200에는 국제 표준에 규정된 공정 파라미터 모니터링 시스템이 장착되어 있습니다. 씰링 로그는 문서화 인터페이스를 통해 발행됩니다.
의료기기에 대한 요건은 지침 93/42/EEC에 명시되어 있습니다. 지침 1조에 따라 93/42/EEC의 섹션 2가 적용되는 경우 제품은 의료기기에 해당합니다.
인체 내 또는 인체에 적용되는 제품을 설명하는 섹션 2a나 섹션 2b(의료기기 액세서리)에는 씰링 장치 및 그 적용에 대한 설명이 포함되어 있지 않습니다. 분류 기준의 부록 IX에는 씰링 장치에 대한 정의가 없습니다.
장치에 대한 정의를 제공하지 않습니다.
따라서 씰링 장치는 의료기기로 분류되지 않습니다. 그럼에도 불구하고 씰링 장치를 개발 및 생산할 때 MELAG은 지침 93/42/EEC의 요구 사항을 준수합니다.
필수 포장 및 표시를 포함한 멸균 의료 기기의 운송, 시운전 및 보관은 국제 표준에 의해 규제됩니다. 멸균 상태의 손실은 보관 중 외부 영향에 의한 것보다 보관 시간의 길이에 덜 영향을 받습니다,
운송 및 사용. 진료 및 클리닉 사용을 위한 보관 기간을 설정하는 것은 불가능합니다.
의료 기기의 보관 기간과 관련된 권장 사항은 종이 및 투명 포장 또는 기타 이에 준하는 포장으로 서랍, 찬장 등 보호된 보관 장소에 6개월 동안 보관할 수 있습니다. 기구의 보관 기간은 개별적으로 지정된 유효기간을 초과할 수 없습니다.
포장 시스템(멸균 차단 시스템과 보호 포장의 조합)은 제조업체가 다른 유효기간을 정하지 않은 한 5년 동안 보관할 수 있습니다.
포장되지 않은 기구는 24시간에서 48시간 이내에 사용해야 합니다. 항상 해당 국가의 요건을 고려하세요.
의료 기기를 보관하는 공간은 건조하고 어둡고 서늘하며 청소하기 쉬운 곳이어야 합니다. 일상적인 활동에서 접근이 불가능해야 합니다. 국제 표준 요건을 준수하는 방과 찬장에 기기를 안전하게 보관할 것을 권장합니다.
멜라폴은 국제 표준을 준수하며 멜라그 밀봉 장치에 완벽하게 적합한 투명 멸균 포장(롤 또는 다양한 크기의 백으로 제공)입니다. MELAfol은 찢어지지 않고 세균으로부터 밀봉되며 쉽게 개봉할 수 있습니다. 멸균 및 승인 후 안전한 기기 보관을 위한 MELAfol 제품에 대해 자세히 알아보세요.
이 동영상을 재생하려면 YouTube의 쿠키 및 기타 외부 서비스를 허용해 주세요.