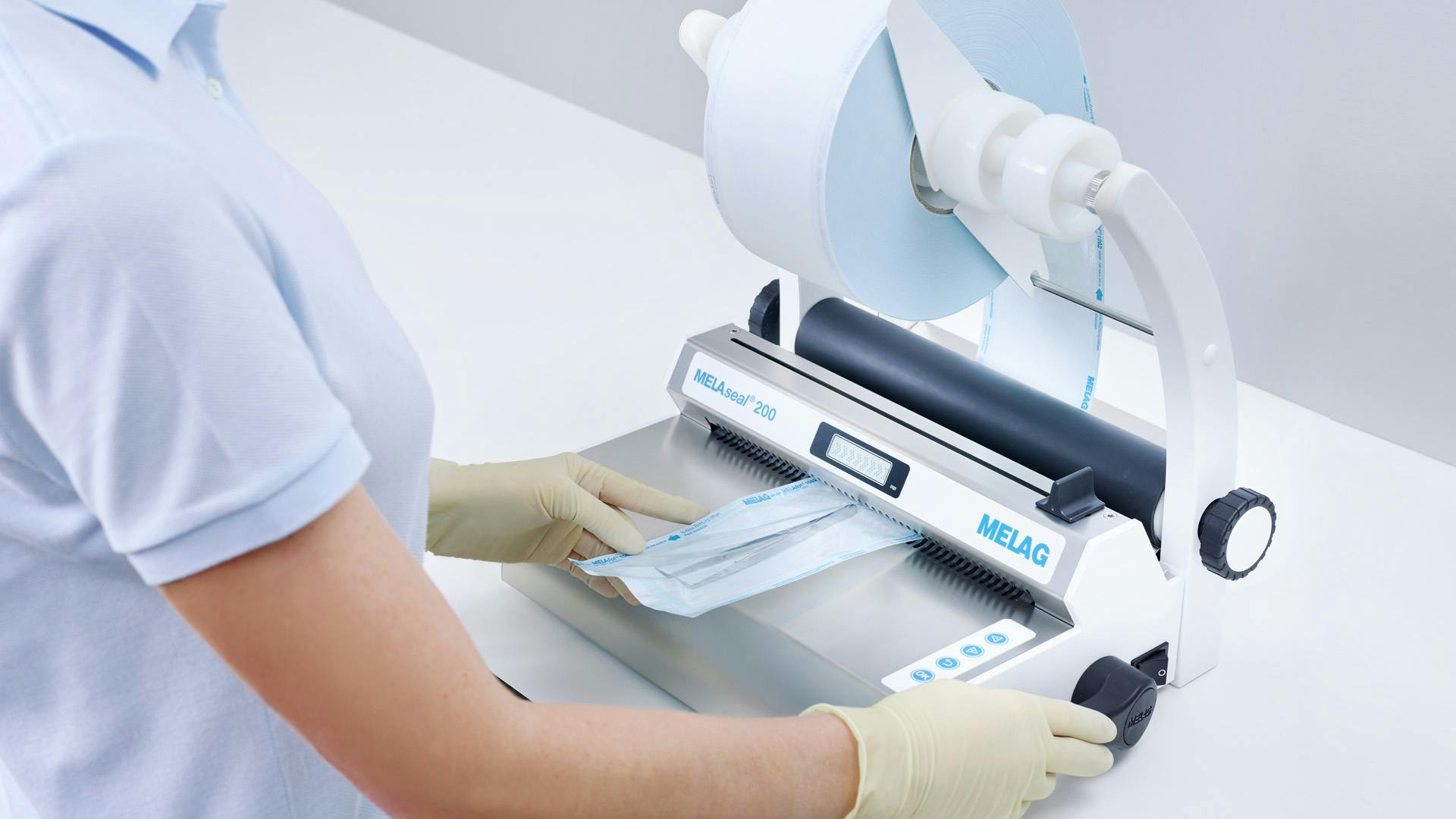
在开始工作前,通过目视和机械检查,每天进行一次密封接缝测试和检查。进行剥离测试时,将密封薄膜包装放入高压灭菌器中灭菌。从蒸汽灭菌器中取出后,顺着剥离方向慢慢拉开密封接缝。目测是为了检查薄膜是否完全脱离纸张。打开薄膜包装时,折痕不得超过 10 毫米。
以可验证的方式记录剥离测试和所有进一步例行检查的结果。我们为此提供了美拉格模板检查表。
可以使用美拉格密封检查仪对密封缝进行日常密封检查。MELAG Seal Check 是一种安全有效的封口检查方法。打印样本上的对比度使实践团队能够快速轻松地确定薄膜层是否与纸面正确融合。将密封检查插入灭菌袋中,如果完整的密封接缝有均匀的变色和清晰的轮廓,则密封检查成功。为了对密封设备的常规测试进行可追溯记录,我们提供了 MELAconnect 应用程序。该应用程序可帮助您通过智能手机或平板电脑上的摄像头轻松将密封检查存档。
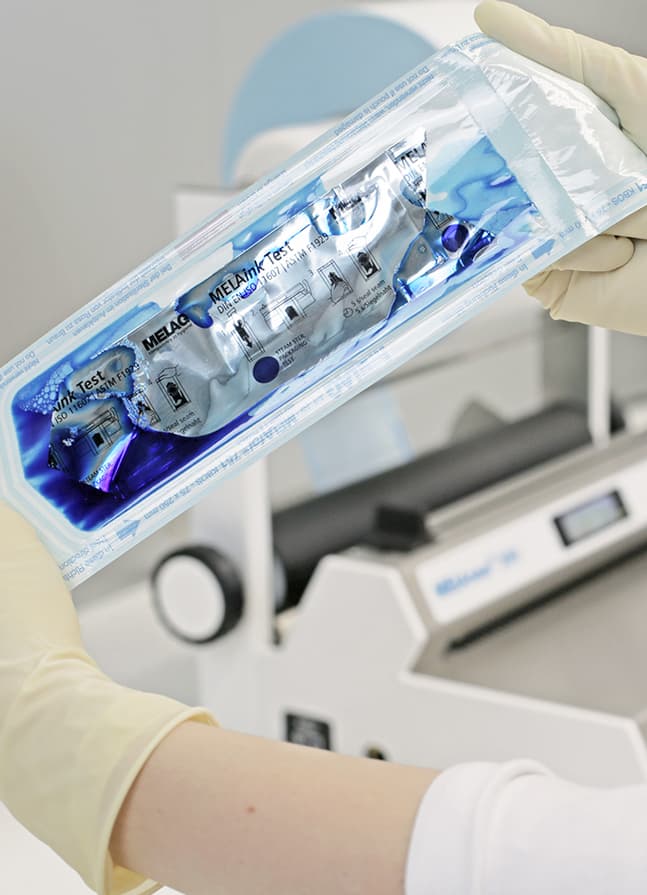
每周一次的 MELAink 测试能更好地识别封口接缝处可能存在的异常。将油墨测试放在一个透明的灭菌包装中并密封。然后将油墨从测试袋中沿着密封接缝的方向压出。墨水的扩散会对包装的所有四个面进行测试,从而发现密封接缝处的不规则、缺陷或通道。
封缝稳定性测试用于对封缝的拉伸强度(抗撕裂性)进行年度检查,并对使用美拉格密封设备的包装过程进行验证。封缝稳定性测试是根据国际标准对您的密封设备进行性能鉴定的标准化测试程序。该测试使用高科技电子测试机进行,确保测试结果有效。下载美拉格密封条接缝稳定性测试申请表。该表格描述了进行密封接缝稳定性测试的所有必要步骤。
要播放此视频,请允许 YouTube 的 Cookie 和其他外部服务。
密封装置可使灭菌器械在病人治疗需要前无污染地存放。我们提供常规测试,如 "密封接缝稳定性测试 "或 MELAink 测试,以检查包装密封接缝的可靠性和安全性。这些测试应按照地区当局规定的时间间隔进行。
长期以来,医院部门一直使用在密封过程中进行自动检测的密封装置,记录温度、接触压力和密封时间这三个工艺参数。目前,许多国家并未强制要求在医疗实践中使用此类可验证的密封装置,但经常推荐使用。
不过,考虑购买新设备的医疗机构和诊所应考虑选择可验证的密封设备,通过对所有工艺参数的持续监控和记录,确保可重复的安全性。
美莱格的密封设备 MELAseal Pro 和 MELAseal 200 配备了国际标准规定的过程参数监测系统。密封记录可通过文档接口发布。
93/42/EEC 号指令规定了对医疗器械的要求。根据该指令第 1 条,如果 93/42/EEC 第 2 节适用,则该产品属于医疗器械。
第 2a 节(描述应用于人体或人体上的产品)和第 2b 节(医疗器械配件)都不包含对密封装置及其应用的描述。分类标准的附录 IX 没有提供密封装置作为医疗器械的定义。
的定义。
因此,密封装置不属于医疗器械。尽管如此,美拉格在开发和生产密封装置时,仍以 93/42/EEC 指令的要求为导向。
无菌医疗器械的运输、调试和储存,包括必要的包装和标识,都有国际标准的规定。无菌状态的丧失与其说取决于储存时间的长短,不如说取决于储存过程中的外部影响、
运输和使用过程中的外部影响。目前还无法确定实践和临床使用的储存时间。
有关医疗器械储存期限的建议是:纸质透明包装或其他同等包装可在受保护的仓库(抽屉、橱柜等)中储存 6 个月。器械的储存期限不得超过单独规定的有效期。
包装系统(无菌屏障系统和保护性包装的组合)可储存 5 年,只要制造商没有确定其他有效期。
打开包装的器械应在 24 小时和 48 小时内使用。请务必考虑本国的要求。
用于存放医疗器械的房间应干燥、阴暗、凉爽且易于清洁。必须避免日常活动。我们建议在符合国际标准要求的房间和橱柜中对器械进行保护性存放。
MELAfol 是一种符合国际标准的透明灭菌包装(有卷装和各种规格的袋装),非常适合美拉格密封设备。MELAfol 无撕口、密封防菌且易于打开。了解更多有关灭菌和认证后安全器械储存的 MELAfol 产品的信息。
要播放此视频,请允许 YouTube 的 Cookie 和其他外部服务。